tl;dr: no
I was talking with my grad student, Lauren Kordonowy yesterday about if error correction should be done to reads prior to abundance estimation. I said no, but told her to do the experiment to convince herself. Now: n=1 and all that, but her results are unequivocal. We used Salmon quasi mapping as implemented in v0.6.0, a mammal transcriptome, and about 20M PE reads of length 150bp. We error corrected using RCorrector. I’m posting this here because it might be useful to somebody. Imagine if we all posted these little 1-off experiments to google-findable places (like blogs)..
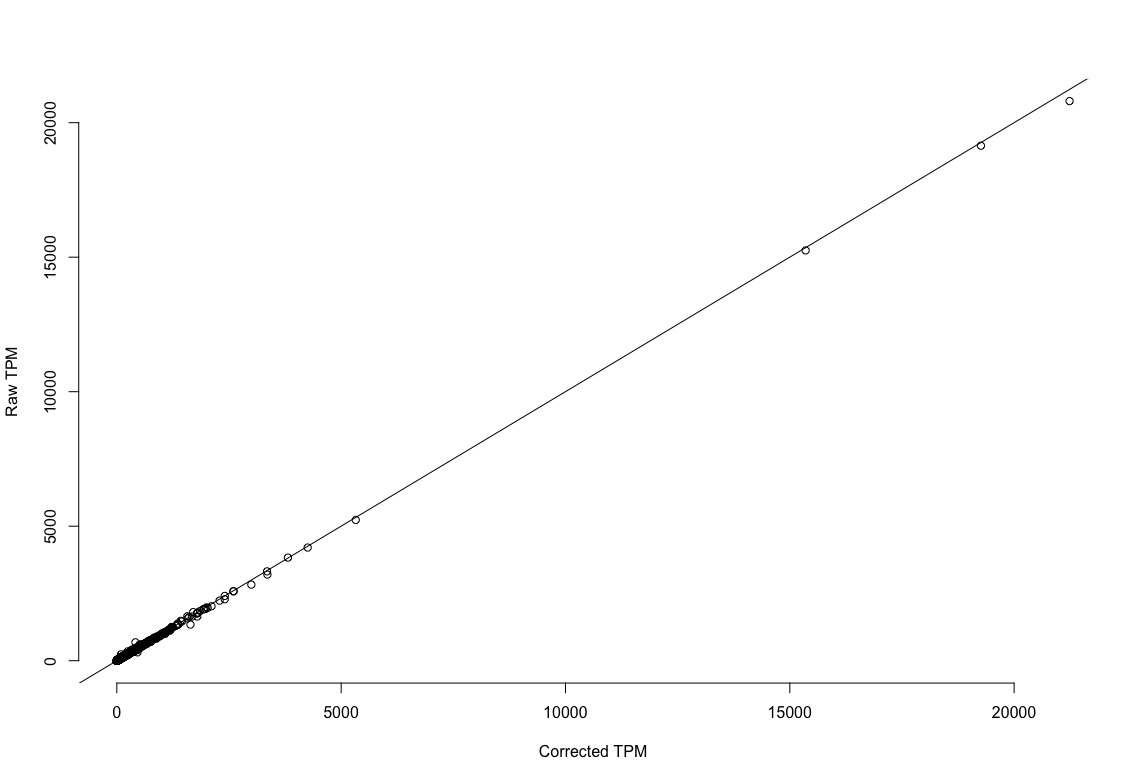
plot1: r^2=.996. The estimates of expression are the same EC reads versus no EC reads!
TPM estimation using corrected reads versus using non-error corrected reads.
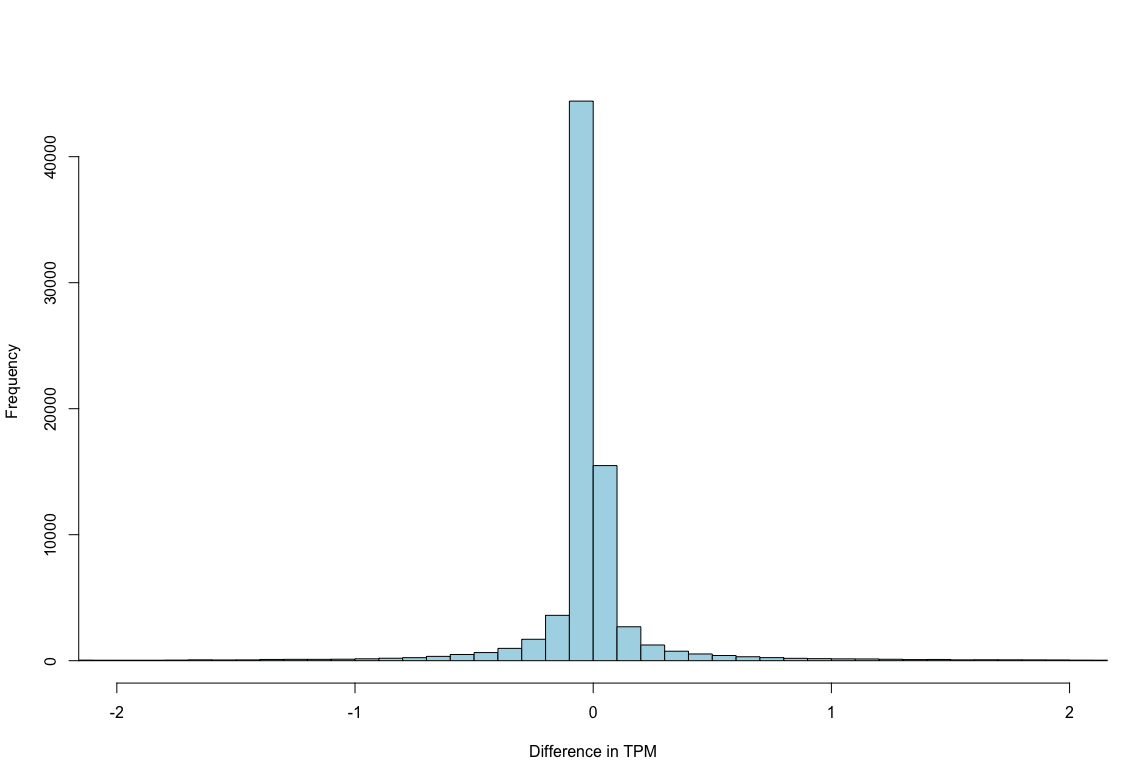
plot2: The distribution of differences in TPM centers on 0.
A niggle
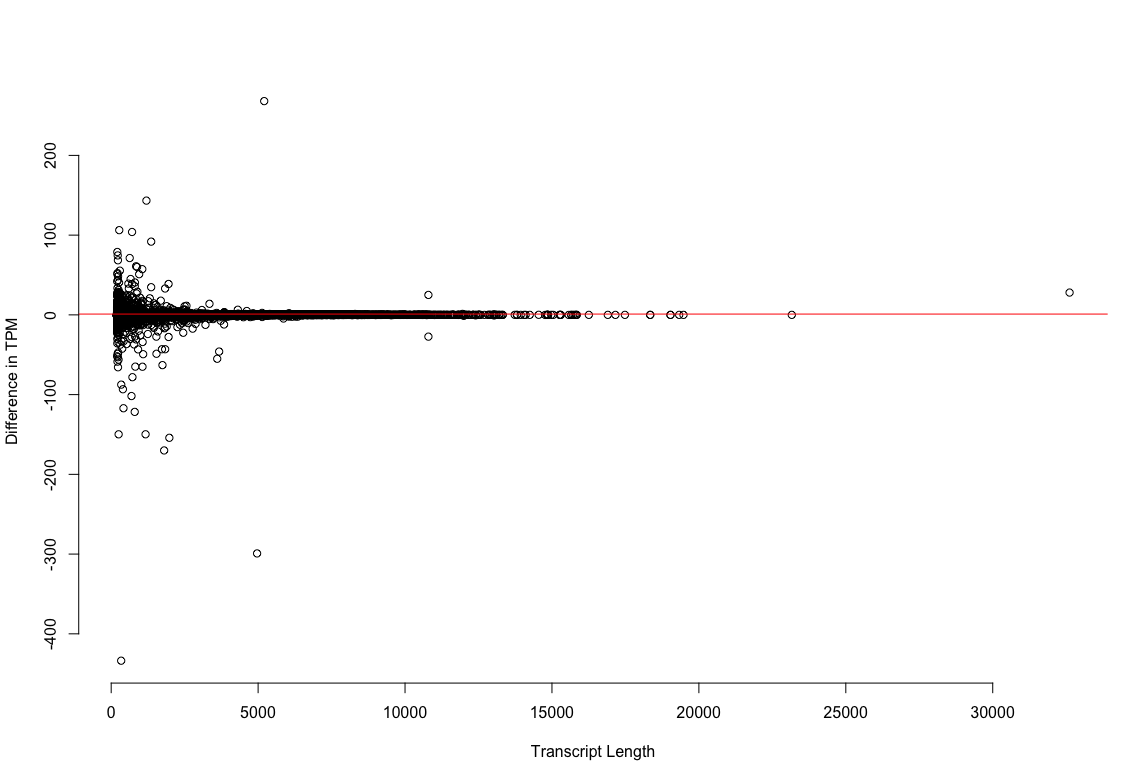
We do see a few hundred transcripts where the correction does change the TPM estimate pretty substantially. What are these transcripts. Are they the short ones, the long ones, the assembly artifacts? Which value for TMP is ‘right’, the one generated using the corrected reads or the uncorrected?
plot3: Estimates of expression for short transcripts (the size class that is enriched for artifactual noise) are maybe* effected by error correction.
*Is this error correction, or just noise in the estimation process. I’m guessing the latter.
So.. don’t bother error correcting your reads prior to abundance estimation.